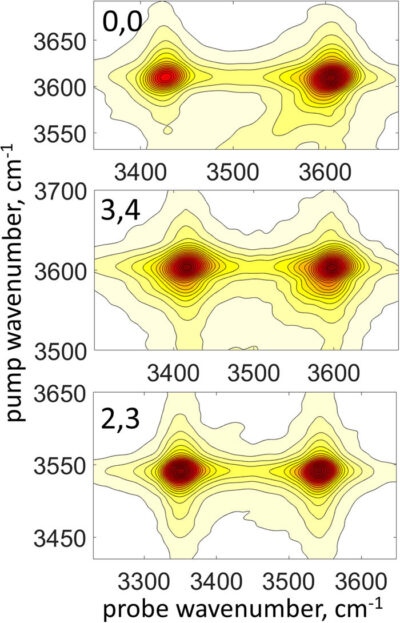
Ultrafast spectroscopy of molecular systems involving hydrogen- (H−) bonding has been at the forefront of fundamental chemical and physical research for several decades. Among the spectroscopic observables of the ultrafast dynamics is the pure dephasing of vibrationally excited molecules. Using third-order nonlinear vibrational spectroscopy, including polarization-selective transient grating measurements of vibrational lifetime and orientational diffusion as well as two-dimensional infrared spectroscopy, we determined different individual line shape components of hydroxyl stretching (νOH) excitations in a homologous series of chlorophenols and obtained the corresponding pure dephasing rates. The pure dephasing rates are correlated with vibrational anharmonicity of the νOH mode, which is tuned remotely from the hydroxyl site by changing the position of the chlorine substituents on the phenol ring. We found that in molecules where the hydroxyl group is in its free form, the pure dephasing rates scale linearly with the mode’s anharmonicity such that assuming it is dominated by the third-order diagonal term, the ultrafast dynamics follow the prediction of the Kubo–Oxtoby theory. However, in the intramolecularly H-bonded ortho-chlorophenols, this trend is reversed, and the pure dephasing slows down by ∼50% for an increase in anharmonicity of only a few wavenumbers. Because the νOH mode’s anharmonicity is known to reflect the H-bonding strength, our results suggest that intramolecular H-bonding can serve as a mechanism of protection from fluctuating forces exerted by the solvent. Such an effect can be relevant for ultrafast dynamics in biomolecules, where H-bonding plays a central role.
Authors: Amit Akiva, Lev Chuntonov
Publication date: 2020/2/18
Journal: The Journal of Chemical Physics
Volume: 152
Issue: 7
Publisher: AIP Publishing